简介
DESeq2 可以利用原始的表达矩阵计算不同处理间的差异表达基因,而获取差异表达基因是转录组分析的第一步。具体讲,DESeq2 输出了两个关键的数据:
- log2foldChange:处理间基因表达变化的倍数的对数 ⇒ 差异幅度
- padj:矫正后的 p 值 ⇒ 结果的假阳性率
这个函数封装了以下 6 步计算,尽管我看不懂:
- estimating size factors
- estimating dispersions
- gene-wise dispersion estimates
- mean-dispersion relationship
- final dispersion estimates
- fitting model and testing
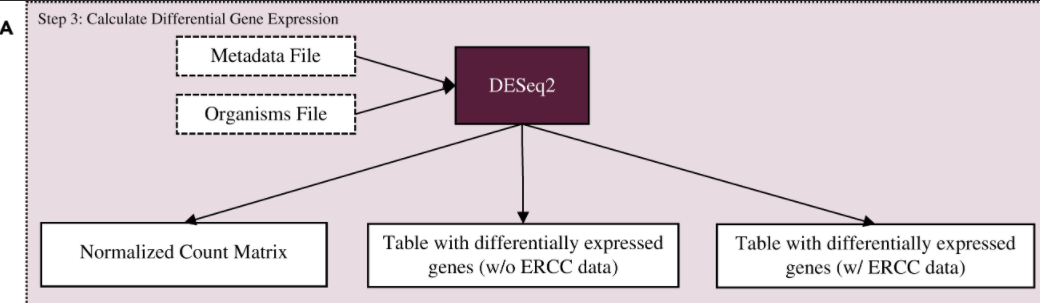
1. 创建 DESeq2 对象
DESeq2 分析需要以下三个数据:
- 原始表达矩阵
- 列信息表,与上一条都在转录组-数据的整理中准备好了
- 实验设计(分析模型)
创建一个 DESeq2 对象可以讲以上三个数据绑定到一起,使分析时的变量管理更加简洁。
其中,分析模型一项是较为重要的。根据具体实验设计和分析方法的不同,可能有较大的差别。在本案例中,使用的是 ~ batch + condition 的方法。其中 batch 为批次,condition 为感兴趣的处理。如此设置可以降低批次效应。
值得注意的,针对不同的检验方法,需要使用不同的对象创建方案,主要是对列信息表和分析模型的改变。注意可以直接对已创建的对象进行修改,这也体现了创建对象的优势。
dds <- DESeqDataSetFromMatrix(
counts_matrix, # 表达矩阵
colData = col_data, # 列信息表
design = ~ replicate + group # 分析模型
)
dds_L <- dds[ , dds$tissue == "L"]
design(dds_L) <- ~ replicate + days # 为 LRT 创建对象2. 运行 DESeq2 主函数
针对以上的对象运行 DESeq2() 主函数,开始计算差异统计。这一步会花费较长的时间。
dds_L <- DESeq(dds_L, test = "LRT", reduced = ~ replicate)
dds <- DESeq(dds)使用以下语句可以查看计算参数
resultsNames(dds)3. 生成标准化的表达矩阵
运行主函数后,便可以得到在不同条件下,基因的表达变化了,这些数据暂时储存在 dds 对象中。然而,此时的表达矩阵仍然是原始的,需要先进行标准化才能在后续的分析中应用。标准化方法一般使用vst(方差稳定化变换)
vsd <- vst(dds, blind = FALSE) # 运行标准化程序,要求考虑实验设计(blind = F)
vst_matrix <- assay(vsd) # 提取标准化后的矩阵
vsd_L <- vst(dds_L, blind = FALSE)
vsd_L_matrix <- assay(vsd_L)4. 提取差异基因
现在需要讲对象中的差异基因提取出来。在提取前,需要明确感兴趣的对比方向,比如组织间对比,长期影响对比。以下代码将主要的 4 中对比方法产生的结果进行了提取:
- 组织间变化对比 ⇒ 11 天-叶 vs 11 天-根
- 超短期处理变化 ⇒ 0 天-叶 vs 1 天-叶
- 短期处理变化 ⇒ 0 天-叶 vs 7 天-叶
- 长期处理变化 ⇒ 0 天-叶 vs 11 天-叶
对于不同实验条件,提取的方法是重复的,主要注意:
results()函数的contrast选项:用于选定比较对象- 直接提取的结果是 DESeq2 的某格式,需要转换为数据框,也可以进一步转换为 tibble
- 按假阳性率(padj)从低到高排序
对于高变基因,通过简单的条件配合 filter() 函数便可以筛选:
abs(log2FoldChange) > 1:表达量倍数 > 1 倍padj < 0.05:显著
## 组织deg提取 ----
resTis <- results(dds, contrast = c("group", "11_L", "11_R")) |>
as.data.frame() |>
rownames_to_column(var = "gene_id") |>
as_tibble() |>
arrange(padj)
degTis <- resTis |>
filter(abs(log2FoldChange) > 1 & padj < 0.05)
## 超短期deg提取 ----
resSrtH <- results(dds, contrast = c("group", "0_L", "1_L")) |>
as.data.frame() |>
rownames_to_column(var = "gene_id") |>
as_tibble() |>
arrange(padj)
desSrtH <- resSrtH |>
filter(abs(log2FoldChange) > 1 & padj < 0.05)
## 短期deg提取 ----
resSrt <- results(dds, contrast = c("group", "0_L", "7_L")) |>
as.data.frame() |>
rownames_to_column(var = "gene_id") |>
as_tibble() |>
arrange(padj)
degSrt <- resSrt |>
filter(abs(log2FoldChange) > 1 & padj < 0.05)
## 长期deg提取 ----
resLng <- results(dds, contrast = c("group", "0_L", "11_L")) |>
as.data.frame() |>
rownames_to_column(var = "gene_id") |>
as_tibble() |>
arrange(padj)
degLng <- resLng |>
filter(abs(log2FoldChange) > 1 & padj < 0.05)