1. 合并每个样本的 counts
使用 awk 命令把每个样品的 counts 进行合并,或者在 R 中使用 left_join()
以下内容以这个GEO数据为示例。
2. 读入原始 csv 文件并观察
将文件下载并放置到当前的 R 工作目录中,使用 tidyverse 中的 read_csv() 可以读入文件,产生一个 tibble。使用 head() 查看结构
raw_counts <- read_csv("GSE283676_combined_Fielder_HC_LC_counts.csv")
head(raw_counts)# A tibble: 6 × 16
...1 X105_0_L X105_1_L X105_7_L X105_11_L X105_11_R X106_0_L X106_1_L X106_7_L X106_11_L X106_11_R X112_0_L X112_1_L X112_7_L X112_11_L X112_11_R
<chr> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
1 TraesCS1A02G000100 4 1 0 1 4 5 5 0 2 4 6 3 1 1 2
2 TraesCS1A02G000200 229 117 83 66 476 295 280 60 117 223 192 175 95 70 253
3 TraesCS1A02G000300 71 49 91 52 231 119 65 43 77 96 122 141 28 27 157
4 TraesCS1A02G000400 398 352 583 347 1457 1036 716 262 430 778 1004 785 313 302 1178
5 TraesCS1A02G000500 0 0 0 0 1 0 0 0 0 0 0 0 1 0 0
6 TraesCS1A02G000600 3 2 3 2 9 5 1 3 4 7 6 5 2 2 8可见表格的结构:
- 第一列:gene_id (字符串)
- 列名:x105_0_L
- x105:生物重复名称
- 0:处理天数
- L:取样部位,L 为叶,R 为根
- 其余:对应基因在每个样本中的 counts
根据后续 DESeq 分析的要求,首先完成以下两项:
- 生成列注释信息:告诉 DESeq2 每个样品名代表了什么处理,并将数据结构转化为因子
- 将数据转化为数值矩阵:DESeq2 需要一个整数的数值矩阵来进行运算
3. 列注释信息生成
一个列注释信息表的结构应该如下:
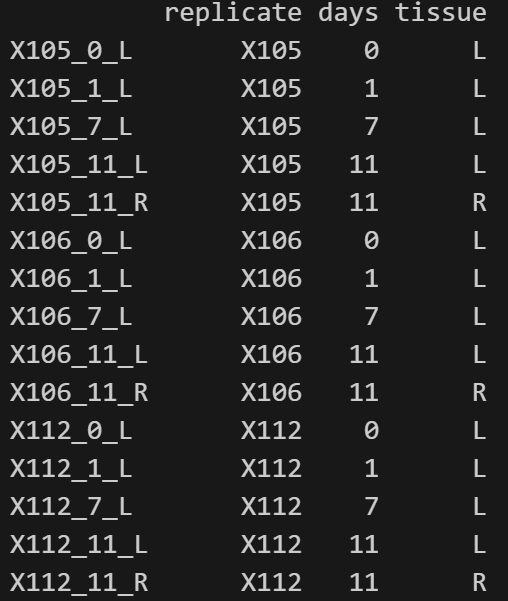
'data.frame': 15 obs. of 3 variables:
$ replicate: Factor w/ 3 levels "X105","X106",..: 1 1 1 1 1 2 2 2 2 2 ...
$ days : Factor w/ 4 levels "0","1","7","11": 1 2 3 4 4 1 2 3 4 4 ...
$ tissue : Factor w/ 2 levels "L","R": 1 1 1 1 2 1 1 1 1 2 ...由于样品名是格式化的且完整储存了所有因子的信息,适当拆分样品名(列名)就可以得到以上表格
代码如下:
library(tidyverse)
raw_counts <- read_csv("GSE283676_combined_Fielder_HC_LC_counts.csv") |> column_to_rownames(var = "...1") # 读入表格并指定基因ID列为行名
sample_names <- raw_counts |> colnames() # 获取列名
col_data <- tibble(sample_names = sample_names) |> # 转换为tibble以供separate()函数使用,命名列
separate(col = sample_names, # 指定需要分割的列
sep = "_", # 指定分隔符
into = c("replicate", "days", "tissue"), # 依次拆分:指定拆分至哪几列中,此处指定了对应列名
remove = FALSE # 不移除来源,即保留分隔前的一列
) |>
dplyr::mutate( # 使用mutate()转换每列成为因子
replicate = factor((replicate)),
days = factor(days, levels = c("0", "1", "7", "11")), # 对于日期,注意指定因子具体值,因为
tissue = factor(tissue)
) |>
column_to_rownames("sample_names") # 指定行名为对应的样品全名
col_data$group <- paste(col_data$days, col_data$tissue, sep = "_")
col_data <- col_data |> dplyr::mutate(group = factor(group)) # 出于实验设计的目的,将“days”和“tissue”合并生成“group因子”数值矩阵的生成
DESeq2 要求传入原始的(counts)表达矩阵,每行为一个基因,每列为一个样本,可以直接进行转换:
raw_matrix <- as.matrix(raw_counts) # 转换为数值矩阵
keep <- raw_matrix |> rowSums() >= 10 # 去除在counts <= 10的基因
counts_matrix <- raw_matrix[keep, ] # 利用上一步的逻辑值向量进行索引其中,去除行和 ⇐ 10 的基因,可以排除在所有样本中都几乎不表达的基因,减轻分析程序的压力。